






 280 / 320
280 / 320

684
En el metabolismo de los INTR no interviene el sistema
enzimático del citocromo P450, por tal motivo son poco
susceptibles de generar interacciones metabólicas rele-
vantes. ZDV y ABC se glucuronidan, por lo que otros
fármacos que afecten la glucuronidación pueden modificar
sus concentraciones. Sin embargo, las interacciones de los
análogos de nucleósidos se deben fundamentalmente a la
potenciación de su toxicidad, por ej. anemia con la asocia-
ción de AZT a ribavirina, cotrimoxazol, o ganciclovir, entre
otros. Lamivudina, emtricitabina, estavudina y tenofovir
se eliminan principalmente por vía renal. Se ha descrito
aumento del riesgo de toxicidad renal al asociar teno-
fovir a algunos inhibidores de la proteasa potenciados con
ritonavir. La combinación de tenofovir con otros fármacos
nefrotóxicos debe evitarse en lo posible (1-3).
Si bien los INTR poseen efectos adversos a corto plazo, hoy
en día denominado “tolerancia”, los efectos adversos más
característicos aparecen a largo plazo y se relacionan con
su toxicidad mitocondrial (4,5). Los mecanismos de toxi-
cidad mitocondrial y celular son complejos, destacando
entre otros el hecho de que estos fármacos, además de
inhibir la transcriptasa reversa del virus, pueden inhibir
la DNA polimerasa gamma mitocondrial. Aunque por su
mecanismo de acción todos los análogos pueden producir
toxicidad mitocondrial, se produce con más frecuencia
con los análogos de timidina, fármacos actualmente en
desuso y por lo tanto, en la práctica clínica estos efectos
son infrecuentes o anecdóticos. Dependiendo del fármaco,
las manifestaciones clínicas pueden ser muy variables:
miopatía, neuropatía, esteatosis hepática y acidosis
láctica, pancreatitis y lipoatrofia periférica (posiblemente
esto ocurra todos los análogos, pero predominantemente
con estavudina y zidovudina) (6,7).
Actualmente se encuentren vigentes 6 fármacos, que por
orden alfabético son: abacavir, didanosina, emtricitabina,
lamivudina, tenofovir y zidovudina.
ABACAVIR (ABC)
Corresponde a un análogo de purina, derivado carbocí-
clico de la desoxiguanosina. Este fármaco presenta un
mecanismo de fosforilación enzimática único, por lo que
es poco probable que compita con la fosforilación de
otros análogos, debe transformarse en carbovir trifosfato,
el cual es el metabolito activo.
Su biodisponibilidad por vía oral es del 83% y presenta
una buena difusión a tejidos, concentrándose en altas
concentración en líquido céfalo-raquídeo (LCR) (alre-
dedor del 30- 40%). Su vía de metabolización es a través
de la glucuronidación y a través de la enzima alcohol
deshidrogenasa. Pese a ello, su interacción con el etanol
no se considera relevante. El resto de sus características
farmacocinéticas se indican en la Tabla 1. Puede tomarse
con o sin alimentos. No requiere ajuste de dosis en insu-
ficiencia renal (Tabla 2).
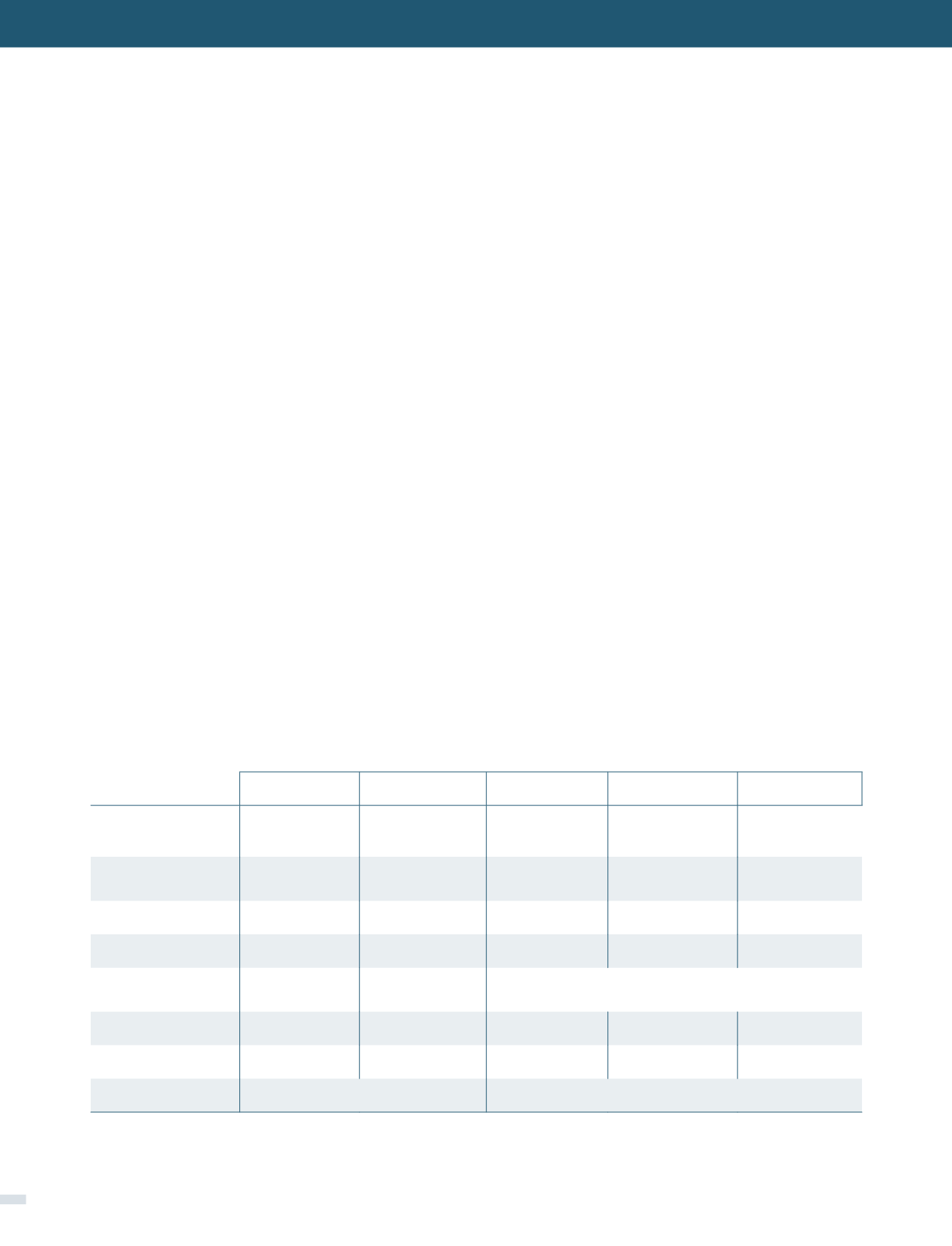
TABLA 1. CARACTERÍSTICAS FARMACOCINÉTICAS INTR (76)
BD: biodisponibilidad, T½p: vida media plasmática, T½ii: vida media intracelular, UPP: Unión a proteínas plasmáticas, LCR: penetración a líquido céfalo
raquídeo.
Ref: Tabla confeccionada con datos extraídos de Micromedex® Healthcare Series y Lexi-comp®.
FÁRMACO
ZIDOVUDINA
ABACABIR
LAMIVUDINA EMTRICITABINA
TENOFOVIR
Dosis normal
300mg/ C 12hr
300mg c/12hr
150mg /c12hr o
300mg/día
No disponible en
Chile
300mg/ c/24hr
BD
64 ± 10%
83%
80-85%
93% comp
75% s.o.
25% ayuna
T ½ p (hr)
0,5 - 3
1,5
5-7
10
12-18
T ½ ii (hr)
3
20,6
16-19
39
10-50
Metabolismo
Glucurónido
Glucurónido,
ALDH
>
70% Renal
UPP
<
38%
49% app
15-36%
<
4%
<
7.2%
LCR
4
3
2
3
1
Actividad
VIH-1,2
VIH-1,2 + VHB
[REV. MED. CLIN. CONDES - 2016; 27(5) 682-697]