








 17 / 164
17 / 164

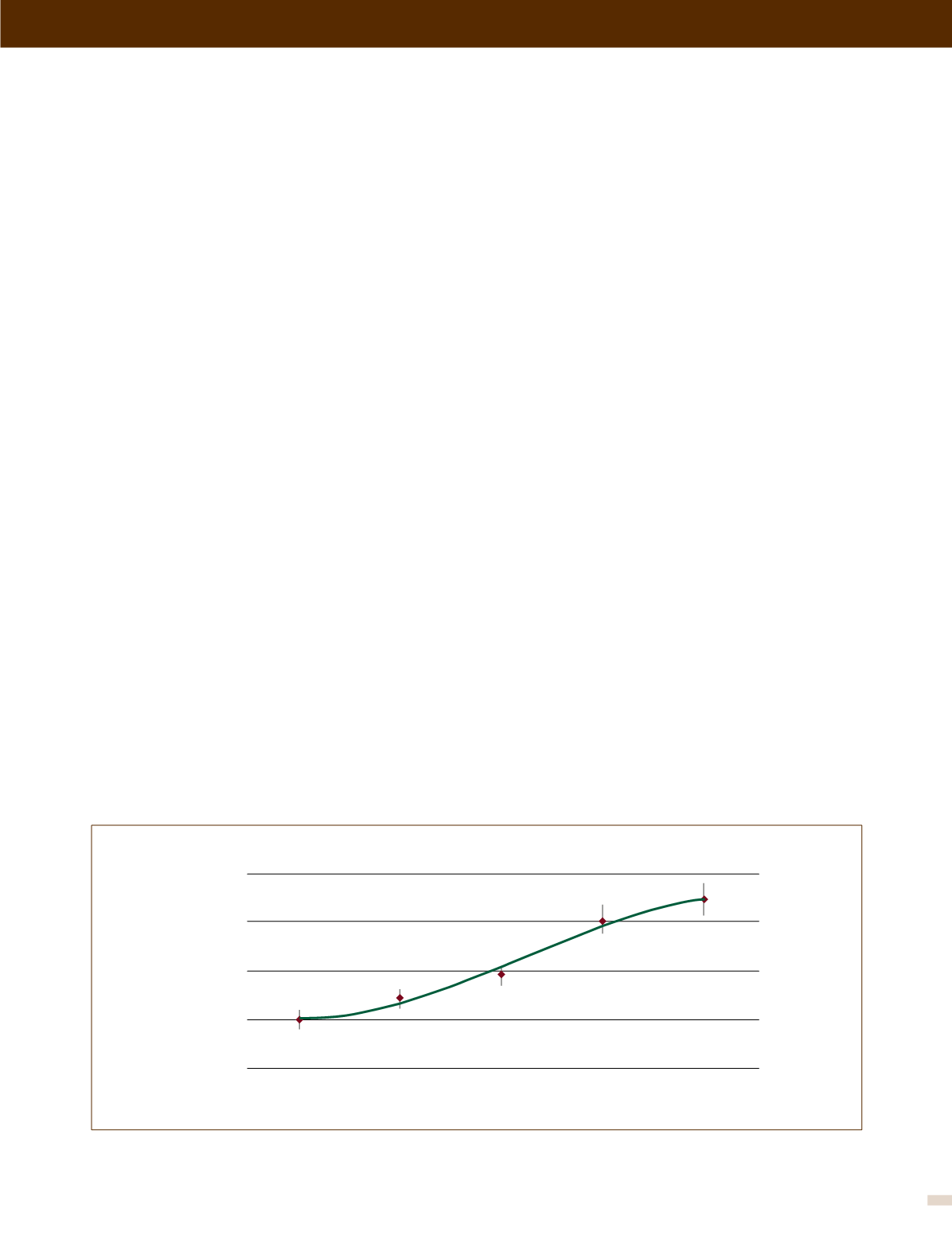
277
Se describen a la fecha más de 2000 mutaciones, siendo la más
frecuente la delta F508. Sin suficientes copias funcionales de la
proteína CFTR en sus membranas celulares, las células epiteliales
no pueden bombear suficiente agua en las secreciones, por lo
que éstas son demasiado espesas y viscosas y tienden a obstruir
los conductos de diversos órganos, especialmente la vía aérea
pequeña en los pulmones.
Esta obstrucción prepara el escenario para la inflamación, la
infección secundaria y la eventual destrucción del tejido que lleva
finalmente a la producción de bronquiectasias que son caracte-
rísticas de la FQ y las infecciones que son la causa final de muerte
de estos pacientes.
El pronóstico de los pacientes con FQ ha mejorado notablemente
en las últimas décadas debido al diagnóstico precoz y a los trata-
mientos médicos que han modificado el curso de esta enfer-
medad, tanto previniendo como retardando sus complicaciones. La
mediana de supervivencia ha pasado de tan solo un año en 1950
a 37,4 años en 2012, según el Registro de Pacientes de la Funda-
ción Americana para la FQ (Figura 1). El resultado es una población
creciente de adultos mayores de 18 años que, en Estados Unidos,
ha llegado a representar el 45% de toda la población con FQ (1).
GENÉTICA
La FQ es causada por una mutación de un gen localizado en el
brazo largo del cromosoma 7 que codifica para una proteína
llamada Regulador de Conductancia Transmembrana de la
Fibrosis Quística (CFTR) (2). Esta, es una proteína que funciona
como un canal que actúa principalmente como un transportador
de cloro. Expresándose en la membrana apical de los epitelios
secretores (pulmón, páncreas, intestino, glándulas sudoríparas,
conductos biliares y conductos deferentes). La alteración de este
canal determina un aumento del cloro en el intracelular y de
una absorción marcada del sodio intraluminal, el que arrastra
agua. Lo anterior produce un espesamiento de las secreciones
de los epitelios comprometidos, con mal funcionamiento de los
cilios y daño en los órganos afectados.
Se describen 6 clases de mutaciones del CFTR:
1- Defectos en la síntesis del CFTR.
2- Defectos en el procesamiento.
3- Defectos en la regulación.
4- Defectos en la conducción.
5- Defecto parcial en la producción o en el procesamiento.
6- Defectos en la regulación de otros canales.
Como el defecto se hereda en forma autosómica recesiva
requiere que ambos padres sean portadores del gen defectuoso y
la probabilidad de tener un hijo es de un 25% con cada embarazo.
MANIFESTACIONES CLÍNICAS
La mutación del CFTR provoca un trastorno en el transporte de
sodio, cloro, bicarbonato que favorece secreciones espesas en el
pulmón, intestino, páncreas, hígado y aparato reproductor que
conducen a su daño (3). Se han descrito más de 2000 mutaciones
y por lo anterior su expresión fenotípica es variable dependiendo
de la o las mutaciones presentes.
40
36
32
28
24
1988-1992
1993-1997
1998-2002
2003-2007
2008-2011
FIGURA 1. Edad promedio de sobrevida, 1998-2012
Referencia 1.
Edad promedio de sobrevida (años)
años
[FIBROSIS QUÍSTICA EN EL ADULTO - Dr. Joel Melo T. y col.]