








 88 / 164
88 / 164

348
El origen es desconocido, pero se postula la existencia de una
predisposición genética sobre la que deben actuar factores
facilitadores y desencadenantes que dan lugar al inicio de la
enfermedad (3). Una base genética poligénica sobre la cual se
superponen factores facilitadores y gatillantes, inflamatorios
y ambientales que permiten el desarrollo y progresión de la
enfermedad (3).
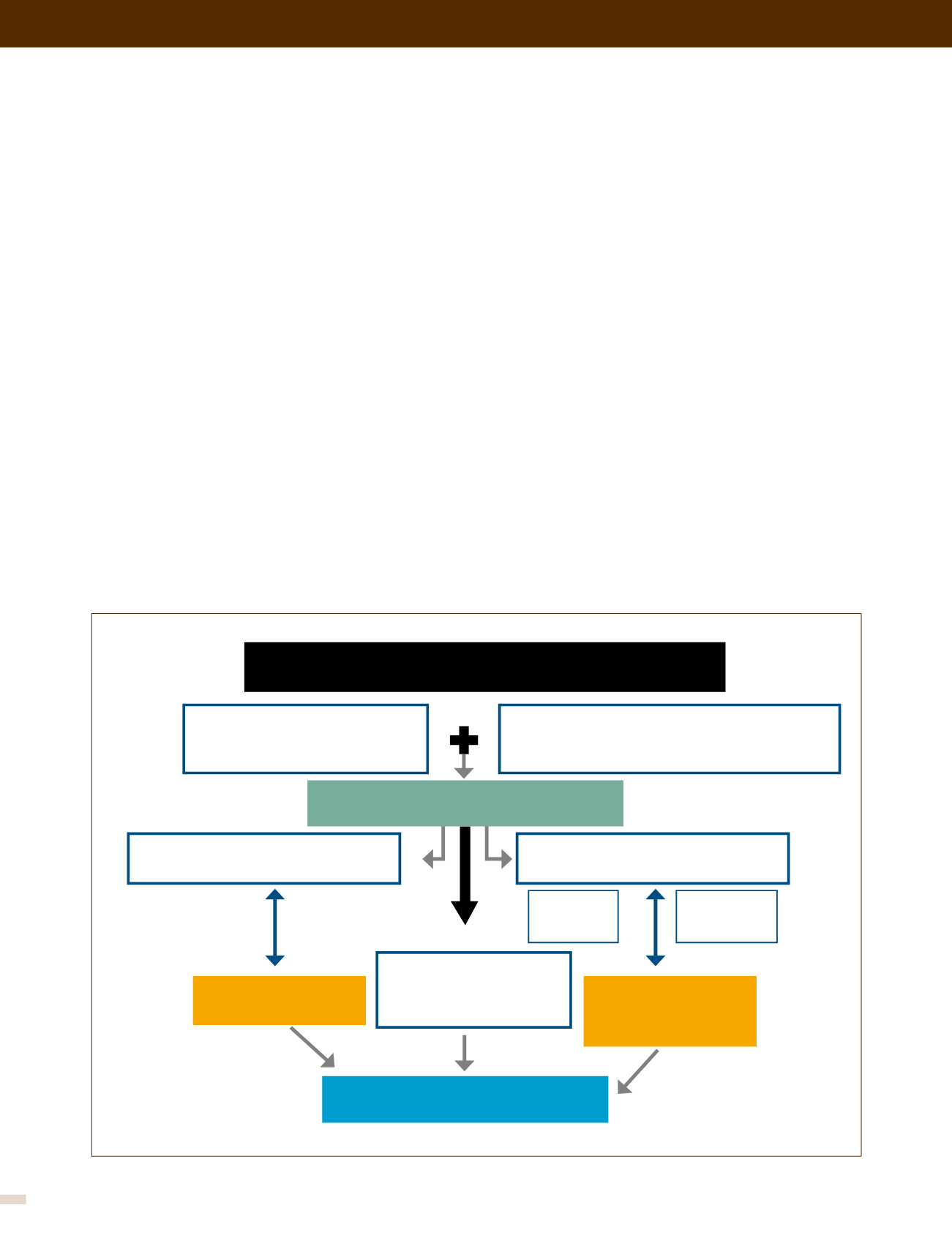
Los diferentes mediadores moleculares implicados en el desa-
rrollo de la enfermedad y los principales mecanismos de acción
y el tipo celular implicado se ilustran en la Figura 1. El efecto
final de estos mediadores es un desbalance hacia los que favo-
recen la vasoconstricción, inflamación, la proliferación celular
y la trombosis vascular frente a los que ejercen el mecanismo
contrario. Se ha relacionado una excesiva vasoconstricción con
la función o expresión anómala de los canales de potasio en
el músculo liso vascular, así como también con la disfunción
endotelial. La disfunción endotelial está involucrada con una
menor producción de agentes vasodilatadores como el óxido
nítrico (ON) y prostaciclina (PG), junto con la mayor expresión
de sustancias vasoconstrictoras y proliferativas como la Endo-
telina (ET) y el Tromboxano A2 (TxA2). El conocimiento de estos
mediadores no sólo es importante para entender la historia
natural de la enfermedad, sino porque son las dianas a las que
se dirigen los diferentes tratamientos actuales y las nuevas
líneas de investigación. En la actualidad se reconocen tres vías
patogénicas, que son además blancos terapéuticos: Vía ON, vía
PG y la vía de la ET (2, 14).
En los últimos años han habido grandes avances en este campo,
fundamentalmente en el estudio de los genes BMPR2 (gen del
receptor tipo II de la proteína morfogenética ósea), ALK1 (acti-
vin-receptor-like kinase1) y 5HTT (endoglina asociada a la telan-
giectasia hemorrágica familiar y el gen del transportador de
serotonina), cuyo polimorfismo LL (dos alelos largos) parece ser
más frecuente en pacientes con HAP que en los controles (3, 14).
El remodelado vascular lleva a una obstrucción progresiva del
lecho pulmonar, con el aumento de la RVP y de la post carga
del ventrículo derecho (VD), que lleva a la hipertrofia y poste-
rior dilatación del VD derecho, lo cual lleva finalmente al dete-
rioro funcional y falla cardiaca derecha (2, 14).
FIGURA 1. DESARROLLO DE LA HIPERTENSIÓN PULMONAR PRIMARIA
HIPERTENSIÓN PULMONAR PRIMARIA
Predisposición genética
BMPR-2,ALK1, %HTT, Eons y
otros
Inflamación - Activación plaquetaria
⇑
Citoquinas,
⇑
serotonina,
⇑
PDGF,
⇓
VIP
Trombosis
in situ
Inflamación
Vasoconstricción
Proliferación
endotelial
Remodelado Vascular Pulmonar
Inicial y progresión
Desbalance
Vasocontricción / Vasodilatación
Hipertrofia Músculo Liso
Vascular
PDGF - VEGF - FGF
Factores de Riesgo
VIH, hipoxia, trombosis, inflamación, entre
otras.
↑
Endotelina
↑
Tromboxano
↑
Serotonina
↓
ON
↓
Prostaciclina
↓
VIP
Injuria vascular inicial
Proliferación y Disfunción Endotelial inicial
[REV. MED. CLIN. CONDES - 2015; 26(3) 344-356]