






 17 / 136
17 / 136

437
padres. Nosotros no recomendamos el estudio cromosómico
si existe antecedente de un hijo con trisomía 13, 18, 21 y la
evaluación del embarazo presente es normal.
Para otras enfermedades o síndromes malformativos infre-
cuentes, es importante conocer la mutación del caso índice y si
se dispone de estudio de ese gen o conjunto de genes causantes
de una enfermedad. En este caso y aconsejando a los padres en
relación a riesgos asociados al estudio invasivo se toma la deci-
sión caso a caso.
IV.- Malformaciones raras con diagnóstico
mediante ultrasonografía y RMF
Entre las malformaciones raras, es decir con frecuencias
menores a 1 en 10.000 recién nacidos vivos diagnosticables en
esta ecografía están:
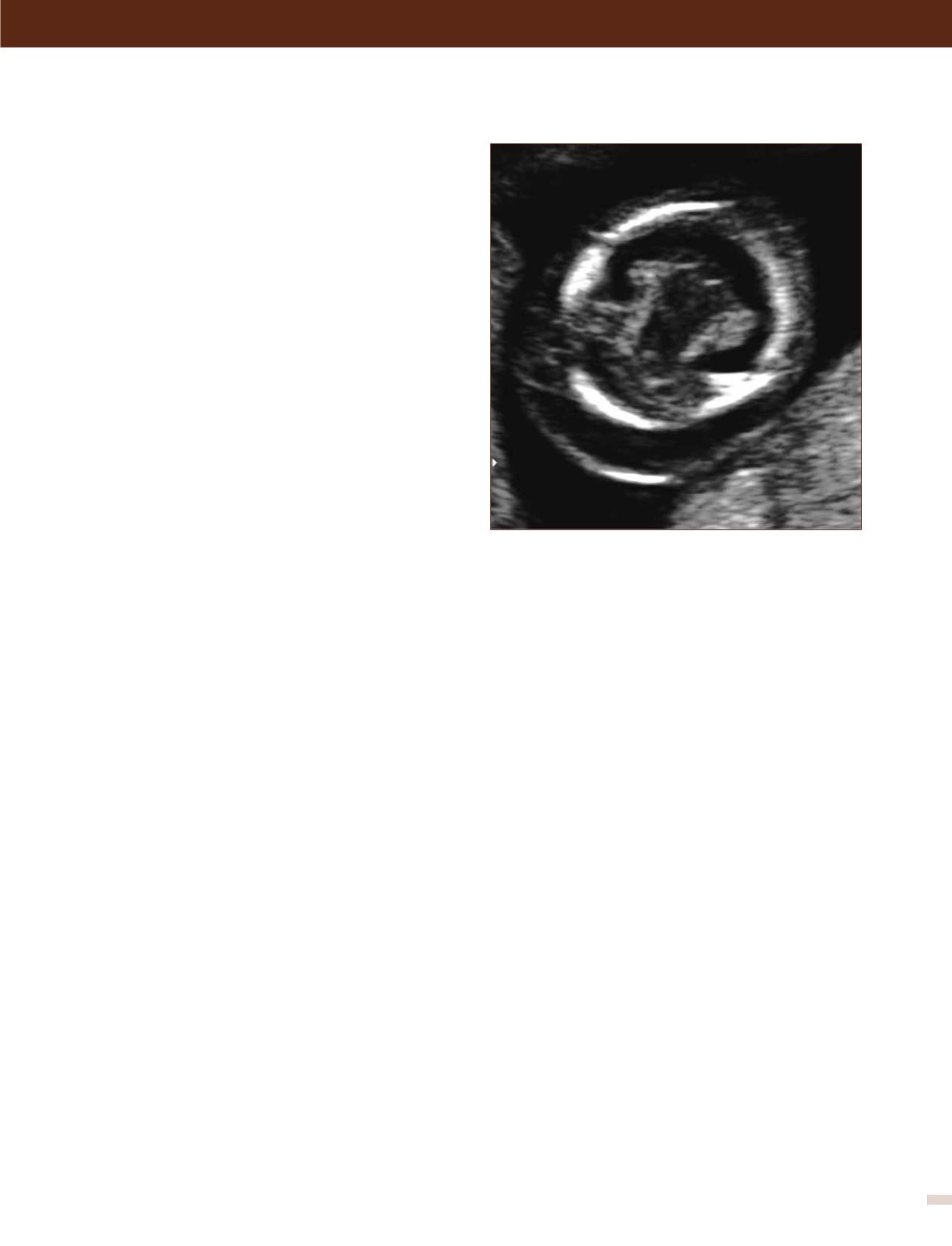
Holoprosencefalia:
Se presenta en 1 de cada 10.000 emba-
razos. Se produce por una alteración en la segmentación del
prosencéfalo en la etapa embrionaria. Su etiología puede ser
de origen cromosómico, (trisomía 13 o 18), genético o en la
mayoría de los casos de etiología desconocida. Se clasifica en
alobar, lobar y semilobar dependiendo del grado de severidad.
En su grado más severo (alobar), el diagnóstico no es difícil y
marca un importante pronóstico fetal. La ausencia de la imagen
clásica descrita como “alas de mariposa” que corresponde a los
plexos coroideos en el plano transventicular del cerebro hace
el diagnóstico a las 11- 14 semanas y la visualización de un
quiste único central sin segmentación central y grados variables
de fusión o segmentación del tálamo con fosa posterior normal
hace el diagnóstico en la ecografía de 20 a 24 semanas.
La holoprosencefalia con defectos faciales se asocia en un 50%
con aneuploidías, particularmente con trisomía 13. Se describen
defectos genéticos de transmisión autonómica dominante y
recesiva así como mutaciones puntuales de algunos genes (SHH,
TGIF, ZIC2 y SIX3). La holoprosencefalia suele formar parte de
los siguientes síndromes genéticos: DiGeorge, Meckel, Kallman,
Smith-Lemli-Opitz, displasia campomelica, Hall-Pallister y
Vasadi. Se han descrito tanto variantes con herencia autonómica
dominante, autonómica recesiva y raramente ligada al cromo-
soma X. El riesgo de holoprosencefalia está aumentado 200
veces en hijos de madres con diabetes insulinodependiente. En
la mayoría de los casos la etiología es desconocida. Para casos
esporádicos, no cromosómicos, la recurrencia clásicamente
reportada es de un 6%. El pronóstico de este defecto es variable
siendo letal para las alobares y semilobar. La holoprosencefalia
lobar se asocia a retraso mental de variable severidad, epilepsia,
espasticidad, distonía y trastornos endocrinos tales como la
diabetes insípida y deficiencia de hormona de crecimiento
(12-19). Figura 4.
Complejo de Dandy-Walker.
Se presenta en 1 de cada 30.000
nacidos vivos. El término Síndrome de Dandy-Walker(CDW) fue
originalmente introducido para indicar la asociación de ventri-
culomegalia de grado variable, cisterna magna aumentada de
tamaño y defecto en el vermis cerebeloso a través del cual un
quiste se comunica con el 4to ventrículo. Con la introducción
de la RMF, apareció el término Complejo Dandy-Walker, que
distingue entre:
a. Dandy-Walker clásico.
Fosa posterior aumentada de tamaño,
agenesia total o parcial del vermis cerebeloso y elevación del
tentorio (Figura 5).
b. Variante de Dandy-Walker.
Hipoplasia variable del vermis cere-
beloso con o sin aumento de tamaño de la cisterna magna.
c. Megacisterna Magna
(habitualmente
>
10 mm). Aumento de
tamaño de la CM con integridad tanto del vermis cerebeloso
como del 4to ventrículo.
Sin embargo y dada la dificultad de encasillar las alteraciones,
éstas deben ser consideradas como parte de un continuo, ya
que en todos los casos es posible encontrar algún grado de
disgenesia del vermis, aún en los casos de megacisterna magna,
mientras que el Dandy-Walker clásico y su variante tienen
tantas similitudes que es imposible a menudo establecer una
distinción precisa. Se asocia a trisomía 13,18 y triploidia, más de
50 síndromes genéticos, infecciones congénitas, teratógenos
como warfarina o puede permanecer inexplicado. El CDW tiene
FIGURA 4. Holoprosencefalia (alobar)
[Diagnóstico prenatal y manejo perinatal en enfermedades raras - Dr. Gustavo Renconret P. y cols.]
Se observa una imagen anormal de los plexos coroideos.