






 42 / 136
42 / 136

462
profundizar en el conocimiento de la variabilidad genética
entre individuos de la especie humana.
Uno de los proyectos más importantes de la última década ha
sido el proyecto de HapMap iniciado en 2002 (27), mediante
un consorcio entre Japón, Reino Unido, Canadá, China, Nigeria
y Estados Unidos. El objetivo de este proyecto fue identificar y
catalogar las similitudes y diferencias de diferentes individuos
de origen africano, asiático y de ascendencia europea, mediante
comparación de sus secuencias genéticas. La identificación de
regiones cromosómicas donde se comparten variantes gené-
ticas permitiría encontrar genes implicados en enfermedades
y variantes asociadas a respuesta a los diferentes fármacos. El
proyecto de HapMap finalizó con la identificación de más de 8
millones de variantes comunes a lo largo del genoma, la mayoría
de ellas localizadas mediante secuenciación Sanger (28, 29) y ha
permitido la comparación de estas variantes con otros proyectos
como 1000 genomas (30).
La llegada de las nuevas tecnologías de secuenciación ha aumen-
tado generosamente la cantidad de datos y la velocidad de produc-
ción de los mismos. El proyecto “1000
genomes
” fue pionero en
emplear la secuenciación masiva de miles de individuos mediante
tecnología NGS (31). Desde su inicio en 2007, este proyecto ha
logrado determinar la localización y frecuencias alélicas de más
de 15 millones de
Single Nucleotide Variants
(SNVs), un millón
de inserciones y deleciones y 20.000 variantes estructurales, la
mayoría de ellas no descritas previamente. Los resultados de este
consorcio estiman que el 95% de la variabilidad de un individuo se
encuentra presente en su base de datos y que cada persona lleva
en su genoma entre 250-300 variantes de pérdida de función en
genes anotados y de las cuales, entre 50 y 100 variantes se han
asociado previamente a enfermedades hereditarias (32).
Durante los últimos años han surgido otras iniciativas interna-
cionales que hacen uso de las nuevas plataformas de secuen-
ciación como el “
International Cancer Genome Consortium
”
(ICGC) (33), el proyecto “
Cancer
Genome
Atlas
” (TCGA) o el
proyecto de ENCODE (34). Este último proyecto ha sido capaz
de dar una imagen muy detallada de todos los transcritos
primarios y maduros, así como la localización de las principales
modificaciones de histonas, sitios de inicio y unión de factores
de transcripción, sitios hipersensibles a DNAsa, descripción de
más de 20.000 seudogenes, modificando el concepto de gen
como la región genómica que codifica un conjunto de trans-
critos alternativos solapantes.
La gran cantidad de información generada por estos y otros
consorcios de investigación ha sido depositada de forma paula-
tina en bases de datos públicas como dbSNP, que han visto
cómo el número de variantes reportadas durante los últimos
años se incrementaba de forma exponencial con la llegada
de las nuevas tecnologías de secuenciación (35), así como la
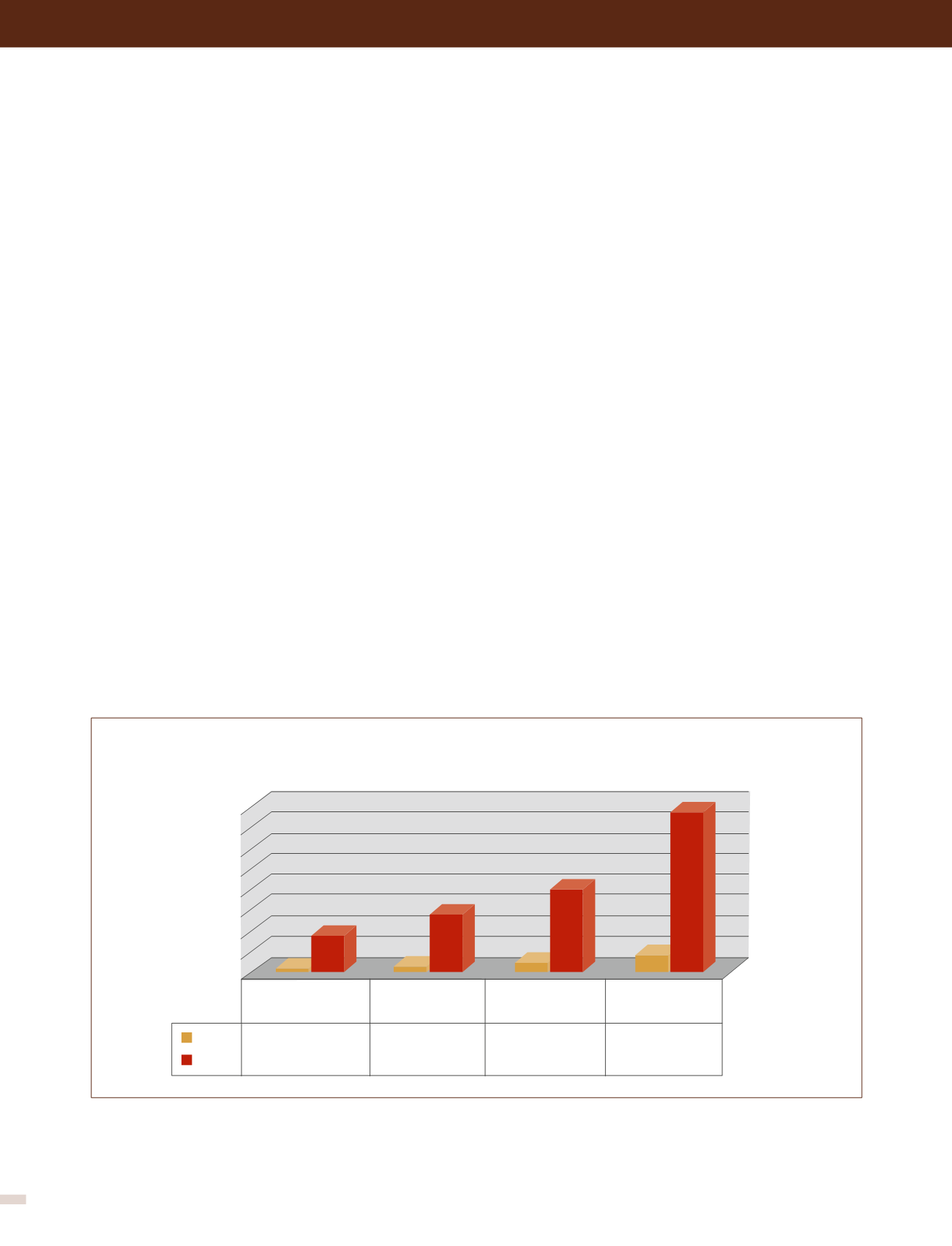
descripción de enfermedades monogénicas (36), Figura 2.
FIGURA 2. Secuenciación masiva en el diagnóstico de enfermedades monogénicas
El incremento exponencial de publicaciones entre 2011 y 2015 revela la ventaja del diagnóstico de enfermedades genéticas a través de la secuenciación
masiva, así como la ampliación del conocimiento científico.
Fuente PubMed. Acceso 2 de mayo 2015.
16000
14000
12000
10000
8000
6000
4000
2000
0
Enfermedades
NGS
Diagnóstico
NGS
NGS en
humanos
877
7988
465
5559
344
3612
2011
2015
1576
15302
NGS global
Incremento Exponencial NGS
2011-2015
[REV. MED. CLIN. CONDES - 2015; 26(4) 458-469]