






 21 / 144
21 / 144

301
De acuerdo al consenso de expertos más reciente,
por definición los cambios neuropatológicos de la EA
necesariamente incluyen depósitos Abeta, con cualquier
combinación de PS y ONF (5). La progresión y el incremento
de la patología de acuerdo a los estadíos descritos están
asociados con una mayor probabilidad de daño cognitivo
de los individuos afectados. En un extremo están los
sujetos presumiblemente cognitivamente intactos, en
una fase 1 de Thal de depósitos Abeta, con bajo estadío
de Braak y escasas PN de acuerdo al CERAD, y al otro
extremo hay casos con extensos depósitos Abeta (fase 5
de Thal), abundantes PN y ONF por toda la neocorteza y
lóbulos temporales mesiales (Braak V o VI) (5).
DEGENERACIÓN LOBAR FRONTOTEMPORAL (DLFT)
La DLFT es un término genérico para el grupo de demencias
degenerativas no-Alzheimer las que se caracterizan por
neurodegeneración concentrada en los lóbulos frontales
y temporales con preservación relativa de los lóbulos
parietales y occipitales. Clínicamente se caracterizan
por trastorno conductual o disfunción del lenguaje,
siendo el trastorno de memoria de aparición más tardía.
La condición clínica se llama demencia fronto-temporal
(DFT) y el sustrato patológico es la degeneración lobar
fronto-temporal (DLFT) (1-3, 10).
Los hallazgos histopatológicos, así como las bases
genéticas que actualmente se conocen, han refinado la
clasificación de este grupo de enfermedades. Actualmente
se caracterizan de acuerdo al tipo de inclusión proteica
encontrada en las neuronas, así como el estatus de
mutaciones, si es conocido (10). Con el progreso de la
investigación en este campo, la clasificación de estas
enfermedades ha estado en un continuo proceso de
actualización y actualmente se basa en el reconocimiento
de alguno(s) de estos tres marcadores demostrados por
la inmunohistoquímica: TAU, en varias combinaciones de
3R/4R (DFLT-TAU); TDP-43, una proteína que une DNA/RNA
(DLFT-TDP-43); y FUS (DLFT-FUS). Dentro de cada uno de
estos grupos, el patrón de distribución de las inclusiones
y los marcadores genéticos son heterogéneos (10). Del
punto de vista clínico los síntomas característicos permiten
formar grupos de DLFT; estos reflejan la distribución
anatómica de la pérdida neuronal más que el tipo de
inclusión. Entre las principales formas de presentación,
las tres mejor caracterizadas son la variante conductual
de DLFT (vcDLFT), la afasia no-fluente primaria progresiva
(APP) y la Demencia Semántica. Es importante destacar
la falta de correlación entre la presentación clínica y
los hallazgos patológicos; por ejemplo, en un estudio
bien caracterizado de sujetos con APP se encontró en la
autopsia que aproximadamente la mitad correspondía a EA
y la mitad tenía una forma de DLFT, la que se dividía más o
menos igual entre DLFT-tau y DLFT-TDP-43 (1-3, 10).
DLFT-tau
Estas enfermedades se definen por la combinación de
degeneración lobar e inclusiones que contienen TAU 3R,
4R o ambas formas. Este grupo incluye la enfermedad de
Pick, las Tauopatías con mutación del gen TAU (MAPT) y
otras Tauopatías sin mutación. Adicionalmente, otras dos
enfermedades que se clasifican primariamente como
Enfermedades de Movimientos Anormales (PSP y DCB)
también pueden tener trastorno cognitivo con atrofia
lobar e inclusiones que contienen TAU (1, 2, 10).
Enfermedad de Pick:
Es una demencia esporádica, que
característicamente comienza en la quinta o sexta década
de la vida. Los cambios conductuales son severos. El patrón
clínico de los síntomas corresponde a la distribución de
las lesiones, las que comprometen los lóbulos frontales
precozmente en el transcurso de la enfermedad.
Macroscópicamente el encéfalo exhibe una severa atrofia
circunscrita y mucho más evidente en los lóbulos frontales;
aún cuando compromete los temporales, típicamente
respeta el tercio posterior del giro temporal superior.
Puede haber compromiso hipocampal y ello es responsable
del trastorno de la memoria. La corteza parietal se afecta
rara vez y la occipital siempre está respetada (Figura 7).
Coincidente con este patrón de compromiso lobar,
hay una mayor dilatación de la porción anterior de los
cuernos frontales y temporales de los ventrículos laterales.
[Neuropatología de las Demencias Neurodegenerativas - Dra. Paulina Arriagada B.]
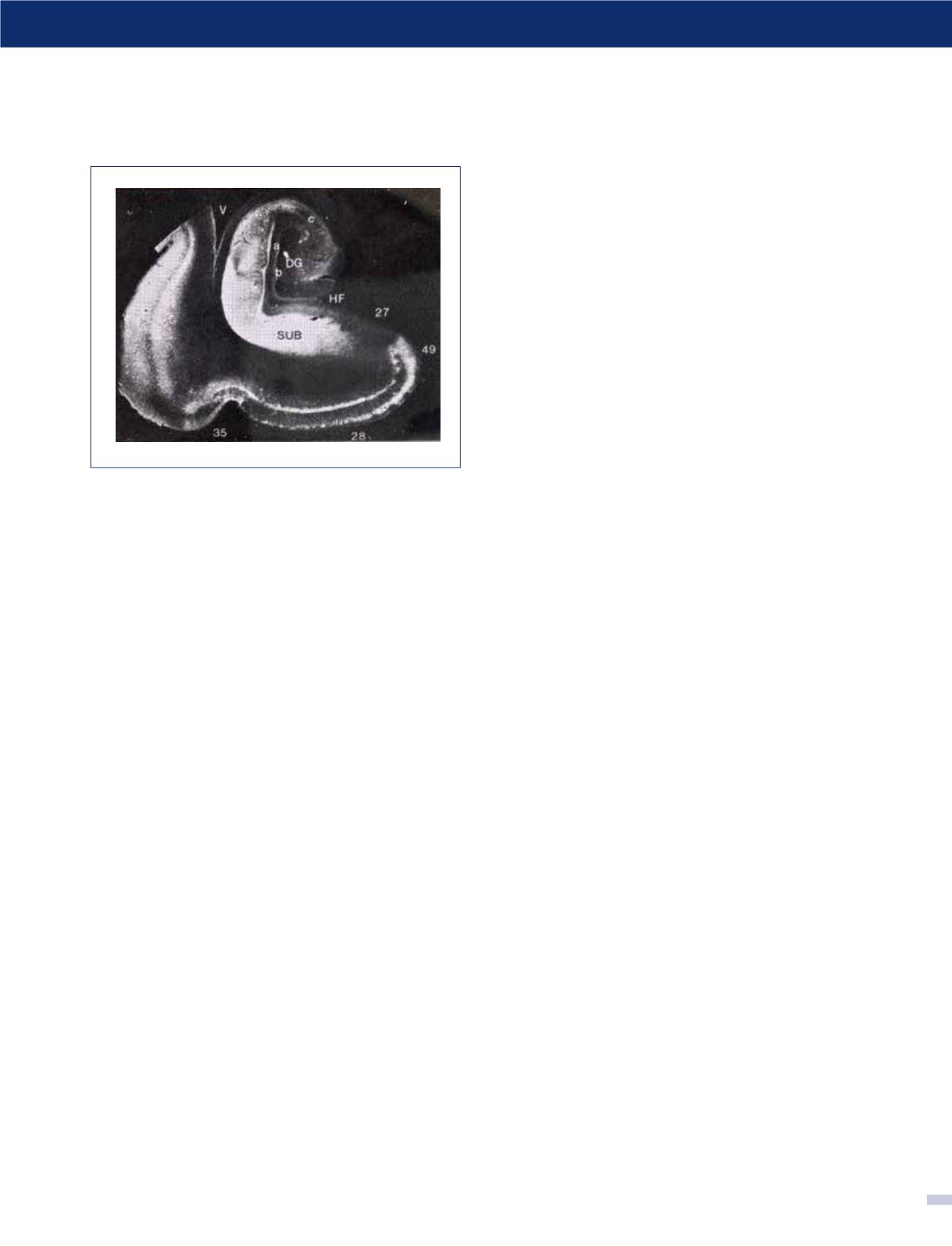
FIGURA 6. Inmunofluorescencia para TAU de la
formación hipocampal en EA
Se destaca la inmunorreactividad positiva de las neuronas vulnerables
en la región del subículum/CAI y capas II y IV de la corteza entorrinal
y neocorteza temporal (estadío VI de Braak). (Foto: colección personal).