






 22 / 144
22 / 144

302
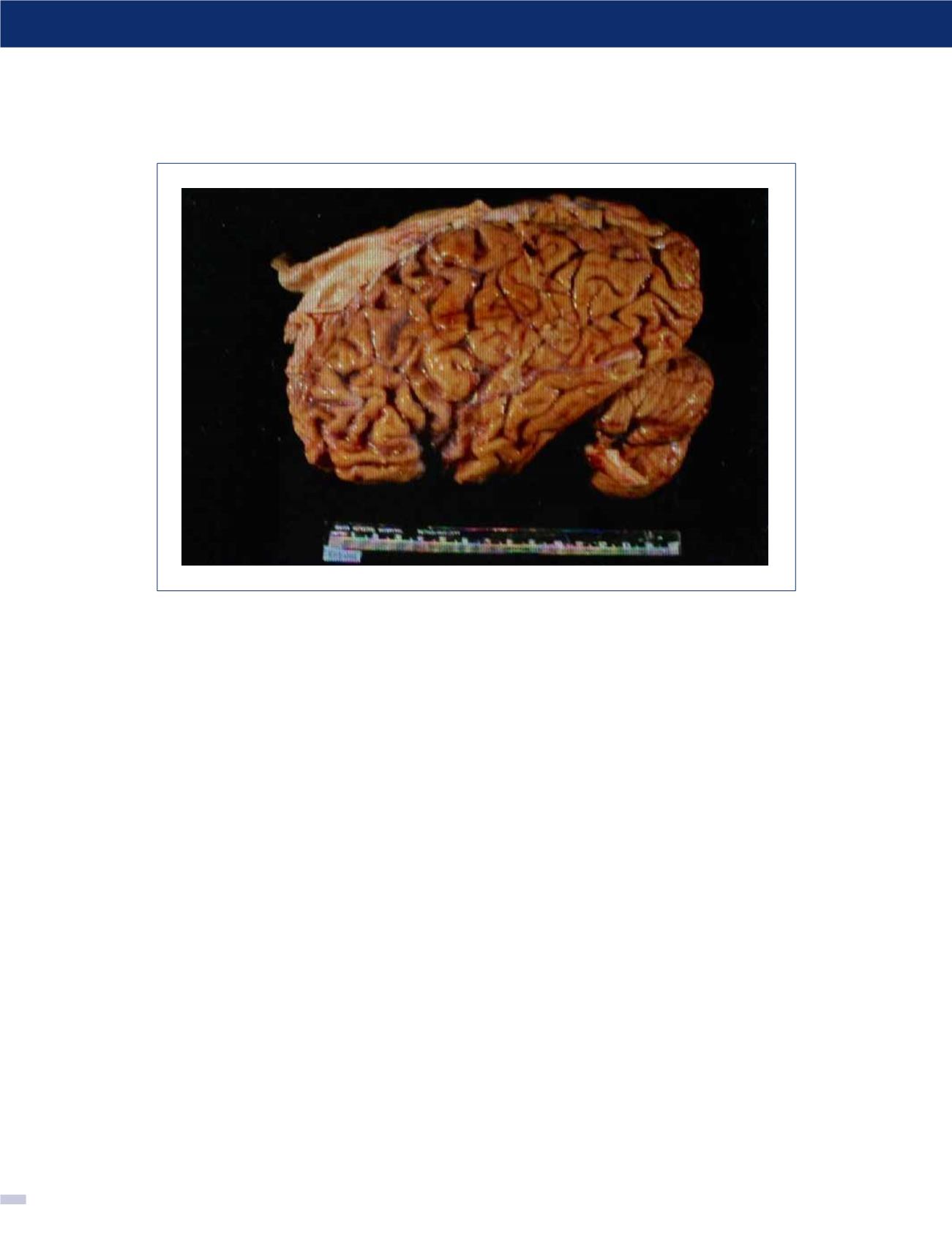
FIGURA 7. Vista sagital de cerebro fresco en la Enfermedad de Pick
Se aprecia la atrofia en los lóbulos fronto temporales, con indemnidad de las regiones posteriores. (Foto: colección personal).
Microscópicamente, las áreas corticales comprometidas
muestran una severa pérdida neuronal, con una densa
gliosis astrocitaria, generalmente acompañada con
microvacuolización de la corteza. La enfermedad de Pick
se caracteriza por la presencia de los cuerpos de Pick
-inclusiones neuronales citoplasmáticas-, redondeadas,
homogéneas, pálidamente visibles con la tinción de H&E.
La inmunohistoquímica muestra que estas inclusiones
contienen 3RTAU. Los cuerpos de Pick son fuertemente
argirofílicos y se detectan muy bien usando las apropiadas
tintines de plata (Figura 8). También son frecuentes de ver
las neuronas balonadas, las que generalmente se conocen
como células de Pick. Cuando están comprometidos los
núcleos grises profundos, también hay importante pérdida
neuronal y gliosis, pero no la acumulación de cuerpos de
Pick. Estos cambios son más predominantes en la cabeza
del caudado, aunque también pueden verse en el putamen,
pallidum y tálamo (1, 2).
DLFT con Mutaciones MAP:
En las formas familiares de
DLFT-TAU se encuentra mutaciones del gen MAPT que
codifica la proteína tau. Estas mutaciones se califican
en aquellas que alteran el acoplamiento del mRNA para
TAU y aquellas mutaciones puntuales. La alteración del
acoplamiento produce una desviación del balance entre el
TAU 3R y 4R. Se piensa que este desequilibrio contribuye
a la iniciación de la disfunción celular. La enfermedad
se caracteriza por atrofia difusa de los lóbulos frontal y
temporales, correlacionada con las alteraciones cognitivas.
Microscópicamente también hay extensa pérdida neuronal
y astrocitosis en las áreas afectadas. Frecuentemente
se encuentra inclusiones TAU en la glía. Las neuronas
acumulan inclusiones TAU en una forma difusa, y se les
llama pre-ovillos. En estas se encuentran isoformas de
TAU3R, 4R o una combinación (Figura 9g y h) (1, 2, 10).
Algunos casos de DLFT no están asociados a patología
de Pick ni mutaciones MAPT, aunque del punto de vista
macro y microscópico no se pueden diferenciar de los
casos con mutaciones.
DLFT con proteinopatía TDP-43 (DLFT-TDP)
La DLFT con inclusiones TAU negativas, TDP-43 positivas da
cuenta de aproximadamente la mitad de los casos de autopsia
de DLFT confirmados. Varios locus genéticos albergan
mutaciones causales para DLFT-TDP e incluye genes para
TDP-43, progranulina y C9orf72. También pueden ocurrir
formas esporádicas. En general, los tipos de inclusiones que
definen los subtipos de DLFT no se correlacionan fuertemente
con la presentación clínica. La DLFT-TDP a menudo se asocia
[REV. MED. CLIN. CONDES - 2016; 27(3) 297-308]